의료기기 관련 자주 묻는 질문
제 제품은 의료기기인가요?
'의료기기'란 제조자가 단독으로 또는 다른 제품과 결합하여 인간에게 다음 중 하나 이상의 특정 의료 목적으로 사용되도록 의도한 모든 기구, 장치, 장비, 소프트웨어, 이식물, 시약, 재료 또는 기타 물품을 의미합니다:
- 질병의 진단, 예방, 모니터링, 예측, 예후, 치료 또는 완화,
- 부상 또는 장애의 진단, 모니터링, 치료, 완화 또는 보상,
- 해부학 또는 생리적 또는 병리적 과정 또는 상태의 조사, 대체 또는 수정,
- 인체에서 유래한 시료(장기, 혈액 및 조직 기증 포함)의 체외 검사를 통해 정보를 제공하는 것, 인체 내 또는 표면에서 약리학적, 면역학적 또는 대사적 수단을 통해 주요 의도된 작용을 달성하지 않지만, 이러한 수단에 의해 기능이 보조될 수 있는 것.
다음 제품도 의료 기기로 간주됩니다:
- 임신 조절 또는 지원 장치;
- 제1조(4)에 규정된 장치 및 본 조항 첫 번째 단락에 규정된 장치의 청소, 소독 또는 멸균을 위해 특별히 설계된 제품.
MDR 제2조를 참조하십시오.
- https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=CELEX%3A32017R0745
- https://health.ec.europa.eu/system/files/2023-09/md_borderline_manual_en.pdf
- https://health.ec.europa.eu/system/files/2023-06/mdcg_2022-5_en.pdf
참고: 모든 수의학 제품은 제외됩니다.
의료 기기의 위험 등급은 무엇인가요?
Class I – 저위험.
EU에서 Class I 의료 기기는 가장 낮은 위험 등급을 갖습니다. 많은 경우 제조사는 통지 기관의 참여 없이 Class I 기기를 자체 인증할 수 있습니다.
이 위험 등급에는 청진기, 붕대, 안경 등이 포함됩니다.
그러나 Class I 의료 기기 내에는 세 가지 추가 하위 분류가 있으며, 이 분류는 위험도가 약간 높으며 제조업체가 CE 마크를 부착하기 전에 지정된 기관의 참여가 필요합니다.
Class I 기기의 하위 분류는 다음과 같습니다 (지정된 기관의 참여가 필요합니다):
Class Is: 의료 기기는 무균 상태로 제공되어야 합니다.
Class Im: 의료 기기는 측정 기능을 갖추고 있습니다.
Class Ir: 의료 기기는 재사용 가능한 수술 도구입니다.
Class IIa – 중간 위험 (NoBo 필요): 이 위험 등급에는 카테터, 보청기, 단기 사용 콘택트 렌즈 등이 포함됩니다.
Class IIb – 중간 위험 (NoBo 필요): 이 위험 등급에는 인큐베이터, 인슐린 펜, 장기 사용 콘택트 렌즈, 인공호흡기 등이 포함됩니다.
Class III – 고위험 (NoBo가 필요합니다): 이 위험 등급에는 심장 박동기, 인공 심장 판막, 수술용 메쉬, 유방 임플란트 및 사용 기간 동안 지속적인 모니터링이 필요한 기타 기기가 포함됩니다.
MDR이란 무엇인가요?
MDR이란 무엇인가요?
MDR은 의료기기 규정(EU 규정 2017/745)을 의미하며, 이전 지침인 활성 이식형 의료기기 지침(90/385/EEC)과 의료기기 지침(93/42/EEC)을 대체하기 위한 정정 및 개정 사항을 포함합니다.
의료기기 규정은 의료기기의 안전성, 품질, 성능을 보장하고 환자 및 사용자의 건강과 권리를 보호하는 것을 목적으로 합니다.
레거시 기기란 무엇인가요?
EU 의료기기 규정(MDR) 및 체외 진단 의료기기 규정(IVDR)에 따른 기존 의료기기는 해당 규정의 적용일(즉, EU MDR의 경우 2021년 5월 26일, IVDR의 경우 2022년 5월 26일) 이후 시장에 출시될 수 있는 기기이며, 각각 EU MDR 제120조 제3항 및 제110조 제3항에 규정된 전환 조치에 따라 적용됩니다.
액티브 기기란 무엇인가요?
'활성 장치'란 인체에서 해당 목적으로 생성된 에너지나 중력 이외의 에너지 원천에 의존하여 작동하며, 해당 에너지의 밀도를 변화시키거나 변환하여 작용하는 장치를 의미합니다. 활성 장치와 환자 사이에 에너지, 물질 또는 기타 요소를 전달하는 데 사용되지만, 해당 과정에서 중요한 변화를 일으키지 않는 장치는 활성 장치로 간주되지 않습니다. 소프트웨어도 활성 장치로 간주됩니다.
제2조
https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:32017R0745
제 기기의 분류는 무엇인가요?
문서 확인을 위한 안내:
- MDR 부속서 VIII에 따른 분류 규칙
- MDCG 2021-24 “의료기기 분류에 대한 지침”
MDA/EMDN 코드는 어디에서 찾을 수 있나요?
MDA/MDN 코드는 장치의 설계 및 목적에 반영되며, 따라서 기술 문서 검토에 참여하는 인력 배정에 주로 관련됩니다. 특정 경우에 한해 NB는 감사 과정에서 제품 성능 및 안전성을 평가하기 위해 제품 검토원을 지정할 수 있습니다.
예시 1: 안과 굴절 수술용 수술용 레이저는 MDA 0302 활성
비이식형 장치로 분류되며, 비이온화 방사선을 사용하는 장치에 해당되므로 MDA 0309 활성 비이식형 안과용 장치에 할당되지 않습니다. 이는 두 코드 모두 해당 장치에 특정적이지만, MDA 0302가 목록에서 더 상위에 위치하기 때문입니다.
예시 2: 정형외과용 나사는 MDN 1102 비활성 골 및 정형외과용 임플란트에 할당되며, 1104 비활성 연부 조직 및 기타 임플란트에는 할당되지 않습니다. 이는 MDN 1102가 목록에서 더 상위에 위치하기 때문입니다.
유럽 의료기기 명명법 (EMDN)
유럽 의료기기 명명법 (EMDN)은 유럽 의료기기 데이터베이스 (EUDAMED)의 운영을 지원하기 위해 마련되었습니다. 다양한 용도 중 하나로서, 제조사는 EUDAMED에 의료기기를 등록할 때 EMDN을 활용하며, 이 과정에서 각 의료기기는 고유 장치 식별자 – 장치 식별자 (UDI-DI)와 연결됩니다. EMDN은 주로 MDR 및 IVDR 요구사항을 지원하기 위한 규제 목적으로 사용되지만, MDR/IVDR 기기 문서화 및 기술 문서화, 지정 기관에 의해 수행되는 기술 문서 샘플링, 사후 시장 감시, 안전성 감시 및 사후 시장 데이터 분석 등에서도 핵심 역할을 합니다. 이 시스템은 MDR/IVDR 하에서 모든 관련 주체가 활동을 수행하는 데 지원을 제공하며, 환자가 자신의 기기 및 EUDAMED에 등록된 시장 내 모든 기기에 대한 핵심 기기 설명을 제공하도록 설계되었습니다.
자세한 정보:
https://health.ec.europa.eu/system/files/2020-09/md_mdcg_2019_14_mdr_codes_en_0.pdf
저는 경제 운영자(Economic Operator)인가요?
'경제적 운영자'란
- 제조업체,
- 승인된 대리인,
- 수입업체,
- 유통업체 또는
- 제22조(1)항 – 시스템 및 절차 패키지 및 제22조(3)항 – 시스템 또는 절차 패키지를 멸균하는 자연인 또는 법인을 의미합니다.
이 중 하나에 해당되는 경우, 귀하는 경제적 운영자입니다.
MDR에 따른 제 의무는 무엇인가요?
MDR의 조항을 참조하십시오.
10 - 제조자의 일반적 의무,
11 - 지정 대리인,
13 - 수입자의 일반적 의무,
14 - 유통업자의 일반적 의무.
MDD와 MDR의 차이점은 무엇인가요?
MDD와 MDR의 차이점은 MDD가 의료기기 지침(Medical Device Directive)을 의미하는 반면, MDR은 의료기기 규정(Medical Device Regulation)을 의미합니다. 이들은 모두 유럽 연합(EU)의 의료기기 시장을 규제하는 법적 틀이지만, 몇 가지 중요한 차이가 있습니다:
- MDD는 지침으로, 각 EU 회원국이 자체 국가 법령에 반영하여 시행해야 하는 규칙의 집합입니다.
- MDR은 규정으로, 국가 법령 없이 모든 EU 국가에서 직접 적용됩니다. 이는 모든 EU 국가에서 규칙이 일관되게 적용되도록 보장하여 규제 프레임워크의 전반적인 일관성과 투명성을 높입니다.
- MDD는 60페이지에 걸쳐 23개 조항과 12개 부속서를 포함하고 있으며, MDR은 175페이지에 걸쳐 123개 조항과 17개 부속서를 포함하고 있습니다.
- MDD는 1993년에 채택되었으며 이후 여러 차례 개정되었습니다. MDR은 2017년 5월에 발효되었으며, 3년의 전환 기간 후 2020년 5월부터 완전히 적용됩니다. 개정으로 인해 MDR은 MDD와 활성 이식형 의료기기 및 체외 진단 의료기기와 관련된 두 개의 다른 지침을 대체합니다.
- MDR은 MDD와 비교해 다음과 같은 변경 사항과 개선 사항을 도입합니다:
- 의료기기의 정의 확대: MDR은 의료기기의 정의를 확대하여 기기의 청소, 소독, 또는 멸균을 위한 제품을 명확히 포함합니다.
- 임상 증거 요건 강화: MDR은 의료기기의 안전성과 성능을 입증하기 위해 더 강력하고 광범위한 임상 데이터를 요구합니다. 특히 고위험 기기에 대해 MDR은 새로운 임상 평가 상담 절차를 수립합니다.
- 사후 시장 감시 및 감시 강화: MDR은 제조업체, 수입업체, 유통업체, 지정 기관이 의료기기의 품질, 안전성, 성능을 제품의 전체 수명 주기 동안 모니터링할 의무를 강화합니다. MDR은 또한 사고 및 현장 안전 개선 조치에 대한 새로운 보고 요건과 시한을 도입합니다.
- 추적 가능성 및 투명성 강화: MDR은 모든 의료 기기에 대한 고유 기기 식별(UDI) 시스템을 도입하여 공급망 전반에서 기기의 식별 및 추적 가능성을 향상시킵니다. MDR은 또한 UDI, 인증서, 임상 조사, 사고, 리콜 등 의료 기기 관련 정보를 저장하고 일반에 공개하는 새로운 EU 데이터베이스(EUDAMED)를 설립합니다.
MDR이 도입한 주요 변경 사항은 무엇인가요?
- 새로운 규정은 기존 규정보다 4배 더 길며 5개의 부속서를 추가로 포함하고 있습니다. 이 규정은 의료기기 지침(MDD)을 대체합니다.
- “안전”이라는 단어는 MDR에 290회 등장합니다. 반면 MDD에서는 이 단어가 40회만 사용되었습니다.
- 새로운 법규에서 사용된 용어의 중요한 변경 사항은 기업들이 제품 포트폴리오를 합리화하고 전사적 영향 평가를 수행하여 필요한 변경 사항을 적용해 준수 요건을 충족해야 합니다.
- 부속서 I, 일반 안전 및 성능 요건은 대부분의 기존 기기(MDD 하에서 CE 마크를 받은 기기)에 적용되어야 할 새로운 조건을 규정합니다. 기존 제품은 새로운 규정에 따라 재인증을 받아야 합니다.
- 새로운 규정은 대부분의 기업이 임상 데이터, 기술 문서, 라벨링을 업데이트해야 합니다.
- 유일한 기기 식별(UDI)이 도입되어 경제 운영자 공급망 전반에서 기기를 추적하는 데 도움을 줄 것이며, 모든 라벨에 표시되어야 합니다.
- MDD의 적용 범위는 의료 목적 외 기기 및 AIMD를 포함하지 않았으나, 이들은 모두 MDR에 포함됩니다.
- 의료 기기의 정의가 확대되어 이전에 규제되지 않았던 일부 의료 목적 외 및 화장품 기기가 포함됩니다. 예를 들어 의료용이 아닌 콘택트 렌즈, 지방흡입 장비, 제모 레이저 등이 포함됩니다.
- 제조업체는 안전성과 성능 주장을 입증하기 위해 더 상세한 임상 데이터를 생성하고 제공해야 하며, 더 엄격한 동등성 기준이 적용됩니다.
- 제조업체는 모든 사고, 부상, 사망 사례를 EU 포털에 보고해야 하며, 이는 관련 데이터를 중앙 집중화하여 환자가 안전 관련 정보에 더 쉽게 접근할 수 있도록 합니다. 사망이나 심각한 건강 악화를 초래하지 않은 사고의 보고 기간은 30일에서 15일로 단축됩니다.
- 전환을 진행 중인 기업은 품질 보증, 위험 관리, 사후 시장 기대치 등 핵심 프로세스를 재검토해야 합니다. 이러한 프로세스는 새로운 요구사항에 부합하도록 재실시하기 위해 세심한 검토, 계획 및 업데이트가 필요합니다.
- 많은 의료 기기의 위험 등급 상향 조정 및 재사용 가능한 수술 기기에 대한 새로운 분류가 도입되며, 이는 통지 기관의 감독을 요구합니다.
CE 마크란 무엇이며, MDR과 어떤 관련이 있나요?
유럽 경제 지역(EEA)에서 의료 기기를 출시하기 전에 해당 기기는 CE 마크를 획득해야 합니다. CE 마크는 법적 제조사가 해당 기기를 평가했으며, MDR 2017/745에 따른 일반 안전 및 성능 요건을 충족함을 나타냅니다. 제품이 지정된 기관의 검사를 받아야 하는지 확인해야 합니다. 이 정보는 해당 제품에 적용되는 관련 법규에서 확인할 수 있습니다. 공인 기관의 참여가 필요한 경우, CE 마크에는 공인 기관의 식별 번호가 동반되어야 합니다. CE 마크와 식별 번호는 서로 명확히 연결되어 표시되는 한 별도로 부착될 수 있습니다.
제품을 인증할 수 있는 공인 기관을 검색하려면 Nando 데이터베이스를 사용할 수 있습니다. https://webgate.ec.europa.eu/single-market-compliance-space/#/notified-bodies
제품이 독립된 기관의 검증을 필요로 하지 않는 경우, 기술적 요구사항을 준수하는지 확인하는 것은 귀사의 책임입니다. 이는 제품 사용 시 발생할 수 있는 위험을 추정하고 문서화하는 것을 포함합니다.
CE 표시 – 인증서 취득, EU 요구사항 - Your Europe (europa.eu).
법적 제조업체는 모든 관련 EU 요구사항과의 적합성을 확인해야 하며, 이를 EU 적합성 선언서(DoC)에 명시해야 합니다.
제조업체는 MDR 준수를 어떻게 보장할 수 있나요?
의료기기 제조업체가 MDR 하에서 부담하는 책임의 전체 범위를 이해하는 중요성.
제10조 MDR은 MDR 하에서 모든 의료기기 제조업체 및 기타 경제 주체에 적용되는 일반적인 요구사항을 규정합니다.
제10조는 모든 제조업체가 다음을 준수해야 한다고 규정합니다:
- 의료기기가 MDR에 따라 설계 및 제조되었는지 확인해야 합니다
- MDR 부속서 I 제3조에 따른 위험 관리 시스템을 갖추어야 합니다
- 모든 기기에 대한 임상 평가를 수행해야 합니다
- 최신 기술 문서를 유지해야 합니다
- 규제 준수 책임자(PRRC)에 접근할 수 있어야 합니다
- 품질 관리 시스템(QMS)을 구현해야 합니다
- EU MDR 이해
적절한 적합성 평가(지정 기관 참여 포함)를 통해 MDR 준수 여부가 입증된 경우, 제조업체는 의료 기기에 CE 마크를 부착해야 하며, 고유 의료 기기 식별 시스템(UDI) 사용 요건을 준수해야 합니다. 또한, 지정 기관과의 계약 조건에 따라 MDR 준수 상태가 유지되도록 생산 절차가 마련되어 있어야 하며, 변경 사항은 규제 당국에 신고해야 합니다.
MDR 미준수 시 처벌은 무엇인가요?
새로운 의료기기 규정은 유럽 내 시장 투명성, 제품 추적 가능성, 소비자 안전을 개선하는 것을 목표로 합니다. 규정 미준수 의료기기를 시장에 출시하면 다양한 위험에 노출될 수 있습니다.
- 재정적 손실
- 특히, 규정 미준수로 인한 재정적 손실은 심각한 영향을 미칠 수 있습니다. 당국으로부터의 잠재적 재정적 제재는 물론, 제품 리콜과 같은 조치도 상당한 비용을 초래할 수 있습니다. 마지막으로, 소비자가 소송을 제기할 수 있습니다: 이 유형의 사고와 관련된 비용은 상당합니다.
- 브랜드 이미지 관련 위험
- 의료기기(MD)의 규정 미준수는 기업의 이미지에 빠르게 영향을 미칠 수 있습니다. 언론은 이 유형의 문제를 빠르게 포착하며, 소비자 단체의 증가로 소비자들이 정보를 더 쉽게 빠르게 공유할 수 있게 되었습니다. 이는 물론 최근 몇 년간 디지털 도구와 그 확산에 의해 더욱 용이해졌습니다.
- 회원국의 제재
- 유럽 연합의 회원국은 Regulation 2017/745 준수 위반 시 적용할 처벌 및 제재를 유럽 위원회에 통지해야 합니다. 이러한 제재는 재정적 벌금이나 제품 회수 또는 리콜일 수 있습니다. 2월 말경에 구체적인 내용을 안내드리겠습니다.
MDR은 EU에서 제조되거나 시장에 출시된 의료 기기에만 적용되나요?
MDR은 모든 EU 회원국에 직접 적용됩니다. 따라서 EU 시장 내에서 공정한 경쟁 환경을 조성합니다.
EU 시장에 의료기기를 출시하려는 제3국 제조업체는 의료기기를 EU 시장에 출시하기 전에 해당 규정에서 정한 규칙, 일정, 의무 사항을 숙지해야 합니다.
터키는 이미 EU 의료기기 지침(MDD, IVDD, AIMDD)을 조화시켰습니다. 한편, 터키 의약품 및 의료기기청은 터키의 의료기기 및 IVD 규정을 EU 프레임워크(MDR, IVDR)와 일치시켰습니다. 터키 법은 또한 의료기기 규정으로 불립니다.
MDR에 따른 CE 마크는 영국(잉글랜드, 웨일스, 스코틀랜드)과 스위스에서 인정되나요?
MDR에 따른 CE 마크는 영국(잉글랜드, 웨일스, 스코틀랜드)과 스위스에서 인정되나요? 영국과 스위스는 의료 기기에 관한 자체 법규를 보유하고 있습니다.
GB: 개정된 의료기기 규정(UK MDR 2002) https://www.legislation.gov.uk/uksi/2002/618/contents/made는 영국 시장에서 CE 마크가 부착된 의료기기의 인정을 확대합니다. 의약품 및 의료기기 규제청(MHRA)은 CE 마크가 부착된 체외 진단 의료기기(IVD) 및 의료기기를 영국 시장에 출시하는 시한을 명확히 했습니다. 시한은 지침/규정 하의 기기 분류에 따라 다릅니다. EU MDR에 준수하는 Class I 기기 또는 EU IVDR에 준수하는 Class A 기기는 2030년 6월 30일까지 영국 시장에서 판매될 수 있습니다. Class IIb 기기는 EU에서 연장 조건이 적용되는 경우 2028년 6월 30일까지 CE MDD 인증서를 기반으로 영국에서 판매될 수 있으며, 유효한 CE MDR 인증서를 소지한 경우 2030년 6월 30일까지 판매 가능합니다. 한편, Class III 기기는 EU에서 연장 조건이 적용되는 경우 2027년 12월 31일까지 MDD CE 인증서로 영국 시장에서 판매될 수 있으며, 유효한 MDR CE 인증서를 소지한 경우 2030년 6월 30일까지 판매 가능합니다.
북아일랜드는 의료 기기에 대한 EU 규정을 계속 준수하며, 북아일랜드 시장에 출시되는 기기에는 CE 마크가 필수입니다.
스위스: 스위스는 새로운 EU 의료기기 규정(MDR)과 일치하도록 의료기기 관련 규정을 최근 개정했습니다. 의료기기 규정(MedDO) https://www.fedlex.admin.ch/eli/cc/2020/552/en이 완전히 개정되었으며, 의료기기 시행 규정 1에 보완 조항이 추가되었습니다. 이 새로운 규정은 2021년 5월 26일에 시행되었습니다. 스위스 당국(Swissmedica)은 EU MDR을 충족한 의료기기(CE 마크를 받은 기기)를 인정합니다.
지침에 따라 CE 마크를 받은 의료 기기를 보유하고 있습니다. MDR을 신청해야 하나요?
예, 유럽 단일 시장 및 터키에서 판매를 계속하려면 (영국과 스위스는 특정 규정을 따릅니다). 그렇지 않으면 현재 인증서의 유효 기간이 만료된 후에는 해당 제품을 더 이상 판매할 수 없습니다.
MDR 하에서 지정된 기관(Notified Bodies)의 역할은 무엇인가요?
지정된 기관은 유럽 연합의 관련 당국에 의해 지정된 기관으로, 특정 제품이 시장에 출시되기 전에 해당 제품의 적합성을 평가하는 역할을 수행합니다.
이 기관들은 관련 법규에서 정한 적합성 평가 절차와 관련된 업무를 수행하며, 제3자의 평가가 필요한 경우에 해당됩니다.
Kiwa Medical은 MDR에 대한 다음과 같은 NoBo를 보유하고 있습니다:
- Kiwa Cermet Italy (KCI) (NB0476)
- Kiwa Dare, 네덜란드 (NB1912)
그리고 - 지침 93/42/EEC 및 KCI의 지점
- 키와 벨기에 인증 서비스(Kiwa Belgelendirme Hizmetleri) (NB1984)
Kiwa는 신규 고객을 위한 평가를 수행할 자원을 보유하고 있나요?
MDR에 대한 Kiwa 의료 인증 기관은 고객과의 약속을 이행하기 위해 견적 제공, 기술 검토, 평가, 임상 검토 및 결정 과정 등을 적시에 수행할 수 있는 역량을 유지하기 위해 노력하고 있습니다.
고객 요구사항으로 현지 언어 사용 감사원이 필요한 경우 어떻게 보장할 수 있나요?
현지 평가원 선정 과정은 진행 중이지만, 모든 현지 평가원을 자격을 갖추기 위해서는 NoBo의 시간이 필요할 것입니다.
일부 국가에서는 인건비 문제로 인해 관심이 없을 수 있습니다.
의료기기 경제 운영자는 기술 문서를 영어로 작성해야 합니다. 필요 시 우리 평가원으로부터 번역가를 고용하여 다른 EU 국가에서 평가를 진행할 수 있으며(모든 평가원은 영어를 구사합니다).
모든 기술 문서는 영어로 작성되어야 합니다.
제안서 작성까지의 최대 소요 시간은 얼마인가요?
신청서가 완전히 작성된 후 15영업일 이내에.
계약 체결부터 인증서 발급까지의 소요 시간은 얼마인가요?
Kiwa Medical이 기술 문서를 받은 시점부터 12개월 이상 경과한 후.
(2023년 10월 25일 EU 지정 기관 인증 및 신청 조사(MDR/IVDR)에 따르면: 45%의 지정 기관은 MDR 품질 관리 시스템(QMS) 인증서를 발급하기까지 6개월에서 12개월의 소요 시간을 표시했습니다. 40%의 지정 기관은 MDR QMS 및 제품 인증서를 발급하기까지 13개월에서 18개월의 소요 시간을 표시했습니다. 새로운 마감일이 다가옴에 따라 소요 시간은 더욱 악화될 가능성이 있습니다.)
인증 및 CE 마크의 비용은 얼마인가요?
비용은 제안서에서 상세히 명시될 것이며, 요금은 NoBo의 웹사이트에 공개되어 있습니다.
추천하는 기술 문서 구조가 있나요?
기술 문서는 다음과에 대한 상세한 정보를 포함합니다:
의료기기, 그 용도, 사양, 설계, 제조 과정, 구성, 위험 관리, 제품 검증 및 검증, 시판 후 활동.
기술 문서에 대한 요구 사항은 MDR의 부속서 II 및 III에 규정되어 있습니다.
부속서 II에 따르면, 기술 문서는 다음과 같은 섹션으로 구분됩니다:
- 기기 설명 및 사양(변형 및 액세서리 포함)
- 제조자가 제공해야 하는 정보
- 설계 및 제조 정보
- 일반 안전 및 성능 요구사항
- 위험-이익 분석 및 위험 관리
- 제품 검증 및 검증.
자세한 내용은 Team-NB Position Paper: 의료기기 규정 (EU) 2017/745의 부속서 II 및 III에 따른 기술 문서 제출을 위한 최선의 실천 지침을 참조하세요. https://www.team-nb.org/team-nb-position-paper-on-bpg-technical-documentation/
MDR에 따라 제품 변경을 관리하는 방법은 무엇인가요?
변경 사항이 중대한 것으로 판단되면 해당 NoBo에 연락하여 그들의 지시에 따라 처리해야 합니다.
정식 신청 전 기술/임상 문서를 사전 검토해 주실 수 있나요?
불가능합니다. NoBo로서 우리는 서명된 계약서에 따라 기술 문서를 검토할 수 있습니다.
해당 계약서에 따라 Gap Analysis를 수행할 수 있습니다.
PRRC(규제 준수 책임자)는 무엇인가요?
제15조 MDR은 모든 제조업체가 회사 내 규제 활동에 대한 전담 책임을 지는 PRRC에 접근할 수 있어야 하며, 해당 책임자가 자신의 직무를 수행하는 데 있어 어떠한 불이익도 받지 않도록 보장되어야 한다고 규정합니다. 이는 해당 책임자의 적절한 활동이 회사에 상업적으로 불리한 조치를 초래할 수 있는 경우(예: 안전상의 이유로 기기 시판 중단을 요구하는 경우)에도 적용됩니다.
대부분의 기업은 자체 책임자를 고용해야 합니다.
그러나 MDR은 소규모 제조업체가 자체 인력을 직접 고용하지 않아도 되지만, “영구적이고 지속적으로” 사용할 수 있는 인력을 확보해야 합니다.
UDI(유일한 기기 식별 시스템)는 무엇인가요?
제27조부터 제31조까지의 MDR은 제조업체가 자사 제품이 특정 EU 전체 적용 UDI 시스템에 따라 유일하게 식별될 수 있도록 보장해야 한다고 규정하고 있습니다. 또한 MDR은 제조업체가 자사 제품의 UDI 기록을 보관하도록 요구하며, 고위험 제품의 경우 모든 제품이 판매된 장소에 대한 기록을 유지하도록 규정하고 있습니다.
IFU(사용 설명서)는 무엇인가요?
'사용 설명서'란 제조사가 사용자에게 기기의 목적, 적절한 사용 방법 및 주의 사항을 알리기 위해 제공하는 정보를 의미합니다.
질문이나 신청서를 어디로 보내야 하나요?
인증 및 CE 마크와 관련된 모든 문의나 직접 연락은 다음 이메일 주소로 보내주시기 바랍니다:
MEDICAL@KIWA.COM
이 이메일 주소는 지정된 인력만 접근 가능하며, 귀하의 문의는 Kiwa Medical의 내부 조직에 따라 처리됩니다:
비활성 장치: Kiwa Cermet Italy (KCI)
활성 장치 (전원 공급원이 있는 모든 유형의 장치):
- 네덜란드, 벨기에, 룩셈부르크, 독일, 스웨덴, 핀란드, 노르웨이, 덴마크: Kiwa Dare
- 기타 유럽 국가, 북아메리카, 남아메리카, 오스트레일리아, 뉴질랜드, 아프리카: KCI
- 터키, 중국, 한국, 파키스탄, 인도, 동아시아: Kiwa Turkey (KCI의 지사)
참고1: 지정된 기관의 범위 때문에, MDA 0201
(이온화 방사선을 사용하는 활성 비이식형 영상 장치) 및 MDA 0314 (인간 세포, 조직 또는 장기(체외 수정 및 보조 생식 기술 포함)의 처리 및 보존을 위한 활성 비이식형 장치) 코드에 해당하는 의료 기기는 KCI에서 처리됩니다.
참고2: 고객이 특정 지정 기관을 원할 경우 문제없이 처리되며, 이는 Kiwa Medical에서 내부적으로 처리됩니다.
Kiwa Medical의 정보를 어디서 확인할 수 있나요?
프로세스와 인증 흐름을 이해하기 위한 종합 가이드라인이 있나요?
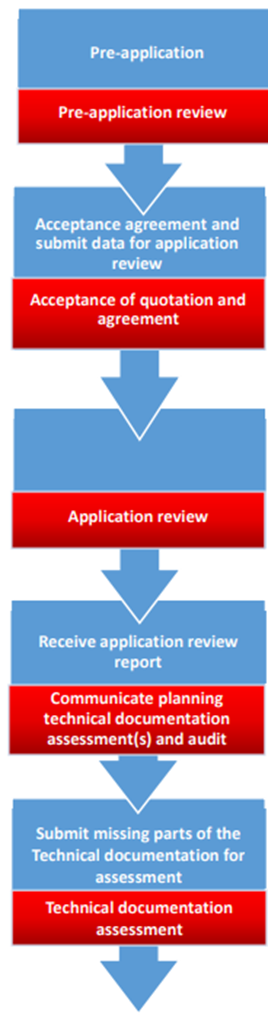 |
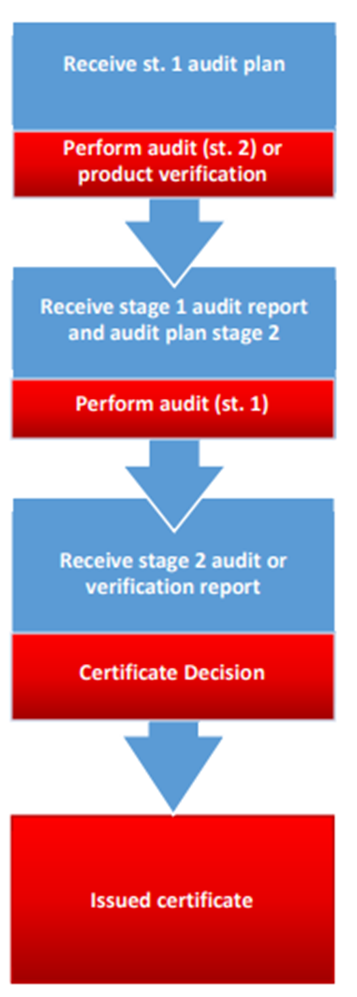 |
문의하기
의료기기 분야에서의 우리 서비스에 대해 더 알고 싶으신가요? 이 양식을 통해 저희에게 연락해 주시기 바랍니다.